Ginkgo.Your Innovation Partner.

One of the challenges we tackle in software at Ginkgo is making critical parts of our apps run faster. Often, this can be solved by upgrading our hardware or by profiling the code and finding obvious slow spots to improve. Sometimes it’s necessary to implement a faster algorithm to get the speed we need. Given the amount of focus algorithms get in computer science, getting to implement one can be both gratifying and a lot of fun. In the following post I will describe how we used a linear-time algorithm for computing the canonical rotation of a string to speed up how we look up circular DNA (circDNA) in our database.
First, some biological context. If a software engineer with no background in biology (which described your humble author four years ago) were to think about how they would represent DNA in a computer program, they would probably want to store it as a string of A, C, G, and T characters. That’s a good first approximation, and it’s basically how we store linear strands of DNA molecules for things like a gene. However, some DNA molecules are circular, which you can imagine creating by taking a linear strand of DNA and joining the end to the beginning, something like this:
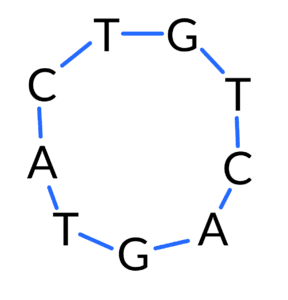
These circular DNA molecules are called plasmids and are usually hundreds or thousands of base pairs long. They are most commonly found in bacteria, but sometimes occur in more complex organisms like yeast or even plants. Addgene has some more information on them. Yeast and bacteria are some of the organisms that Ginkgo engineers modify to produce compounds that we care about, like ones for different fragrances, or ones that help fix nitrogen that helps plants to grow. So we are able to use plasmids in the organism development process, and often do so.
Plasmids are convenient to use, because it’s easier and faster to get them (and their new genes) into an organism than by modifying the organism’s genome, so using them speeds up the development process. They can coexist with genomic DNA in the same organism, and the organism will (usually) express the proteins that the plasmids’ circular DNA codes for, so they are good to test the changes with. However, in most cases, we don’t use plasmids in the final production strain we deliver to customers. We usually encode the final changes in the organism’s genome, since they are easier to maintain in industrial settings. That said, a lot of the DNA molecules that we keep track of at Ginkgo are circular.
The use of circular DNA in the lab brings up an interesting computational problem though, which is how to store it in the database, and how to look it up quickly. (When we query for DNA in this context, it is for an exact match, not a full text search.) An input sequence might have a match in the database, but that match might be rotated a certain amount. For instance, if we had a circDNA sequence of ACGT, we could store it like that, or as CGTA, or GTAC, or TACG. If the sequence is of length n, then there are n rotations of it, and up to n different values for the rotated string. The totally naive solution for looking up a string would be to rotate it n times, and for each rotation, to do a sql query for that rotation of the string. The running time of this solution would be O(n^2).
The solution that we implemented a long time ago, and that served us well for many years, was a special sql query to check if an input sequence was contained in some sequence in the database. The key idea is that if you have a stored sequence of circDNA of length n, you can concatenate it to itself to get a string of length 2*n that contains all the possible rotations of the original. Then you can check if the input value (also of length n) is contained in the concatenated string. If so, then it is a rotation of the stored sequence. For example, if the input data was ATATATAT, and the existing sequences were stored in a field called “data” in a table called “sequences”, the resulting query would be:
SELECT *
FROM sequences
WHERE LENGTH(sequences.data) = 8 AND CONCAT(sequences.data, sequences.data) LIKE ‘%ATATATAT%’This query is correct, and if there are a small number of circDNA molecules in the database with the same length, it also runs quickly. However, if there are m matching sequences of length n in the database, then the running time of this query is O(m*n). Since implementing this approach several years ago, the number of molecules in our database has grown a lot, and for any given length, the odds that multiple circDNA sequences have the same length has grown substantially. Also, last fall we got reports of slowdowns after large new batches of circDNA molecules of the same length were created.
To address this problem, we decided to bring our circDNA storage and lookup solution in line with what we already do with linear DNA. For linear sequences, we store the sequence data as a string, as well as a hash of the sequence string. So linear sequence lookups can be quite fast because we only have to hash the lookup value and then query for the hashed value. Of course, we have a SQL index on the column for the hashed value, which makes lookup even faster.
We wanted to do the same for circular DNA, but then the question arose, which rotation of the string do we store a hash for? An obvious answer was the lexicographically least rotation. For our purposes, “lexicographic” just means alphabetical ordering. For example, although ACGT and GTAC are rotations of the same sequence, A is earlier in the alphabet than G (and every other character in the sequence), so the lexicographically least rotation is ACGT.
As another example, the lexicographically least rotation of GAATAA is AAGAAT. The one-letter prefixes of that sequence are A, G, and T. The two-letter prefixes, in lexically increasing order, are AA, AG, AT, GA, and TA, but there are two occurrences of AA, so we need to consider longer prefixes. The lexicographically least three-letter prefix is AAG, and there is only one occurrence of it, so that’s the start of the canonical rotation.
Fortunately, there is a linear-time algorithm for computing that rotation. In fact, there are a few. We decided to use Booth’s algorithm. The discovery of this algorithm is interesting because it uses a preprocessing method originally used for the Knuth-Morris-Pratt string matching algorithm.
The Knuth-Morris-Pratt algorithm first builds an auxiliary array, in linear time, of the same length as the string to be searched, with certain information about prefixes contained within the string being matched on. The algorithm then iterates over the string to be searched, checking for a match with the input string, and sometimes using the auxiliary array to avoid brute-force string comparisons. Because of how the auxiliary array is built, and used in the search algorithm, the overall time for the search is O(n).
Booth’s algorithm generates the same auxiliary array, but instead of using it to match a string, it uses it to find the lexicographically least prefix of all possible rotations of the string in O(n) time. Since this prefix is the start of the lexicographically least rotation of the sequence, we can treat that rotation as the canonical one.
Over the past couple months we made the necessary code changes to implement and use Booth’s algorithm, and updated the existing circDNA rows in our sequence table with their sequences’ canonical hashes, to be used with the new faster lookup code. Our test benchmarking has shown noticeable improvements in circDNA lookup. In some cases we have seen speedups of 4-6x. These changes are now in production, and we expect circDNA operations to run and scale as smoothly as the ones for linear DNA now. It was fun, and satisfying, to speed up circDNA lookup using the power of a better algorithm!
(Creative Commons Bacterial Plasmid by Spaully is licensed under CC BY-SA 2.5.)
Posted by David Hofer





.svg){kind=link}